CHAPTER 6
Miniprep & NotI digests
PURPOSE
A miniprep is a protocol for isolating plasmids from bacterial cultures. Minipreps from the majority of white clones produced in CHAPTER 5 should result in isolation of BAC vectors containing 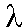 H3 DNA inserts. The pBeloBAC11 vector has a NotI site on each side of the HindIII insertion site. Thus digesting isolated BACs with NotI releases inserts from vector DNA. Pulsed-field gel electrophoresis can be used to check whether white clones are "true positives" (whether they actually contain plasmids with a genomic DNA insert). If numerous false positive clones are observed, the vector was most likely damaged during dephosphorylation and/or the T4 ligase is inactive.
H3 DNA inserts. The pBeloBAC11 vector has a NotI site on each side of the HindIII insertion site. Thus digesting isolated BACs with NotI releases inserts from vector DNA. Pulsed-field gel electrophoresis can be used to check whether white clones are "true positives" (whether they actually contain plasmids with a genomic DNA insert). If numerous false positive clones are observed, the vector was most likely damaged during dephosphorylation and/or the T4 ligase is inactive.
EXPERIMENTAL PROCEDURES
SUPPLIES, EQUIPMENT, AND REAGENTS (see CHAPTER 2 for details): test plates with H3 transformants (see CHAPTER 5); MP-1; MP-2; MP-3; LB+CM; isopropanol (stored at –20ºC); 70% ethanol (-20ºC); 0.5X TBE; PFGE Midrange Ladders or PFGE Lambda Ladder; agarose; NotI with 10X buffer and 100X BSA; blue juice; sterile toothpicks; CHEF gel apparatus; large CHEF gel casting stand (21 x 14 cm); 45-tooth gel comb; UV light box equipped with camera or image-capture system
METHODS:
- For each test plate produced in CHAPTER 5, obtain nine sterile 15 ml culture tubes with caps. Label each set of nine tubes with the Roman numeral designation of one of the test plates (i.e., I, II, III, or IV). In a laminar-flow hood, place 3 ml of LB+CM in each tube.
- Transfer the test plates to the hood. Open a box of sterile toothpicks. Remove the lid from plate I and stab an isolated white colony with one of the toothpicks. Drop the toothpick into an appropriately labeled culture tube. Cap the tube. Pull a second sterile toothpick from the container and pick a second white colony off plate I. Place the toothpick in a second appropriately labeled tube. Repeat this process until you have transferred bacteria from nine different white colonies on plate I to the nine different tubes labeled I.
- Repeat step 2 for each of the other test plates.
- Place the resulting 36 cultures in a shaker incubator at 37ºC overnight (12-18 hours).
Note 6.1: Minipreps can be performed using the protocol below. However, if desired commercially available minprep kits and/or automated DNA isolation systems can be employed to isolate BACs from liquid cultures (see CHAPTER 2).
- Place culture tubes in a swinging bucket centrifuge. Centrifuge at 800 x g for 20 min. Pour supernatants from the tubes.
- Resuspend each bacterial pellet in 200 µl of MP-1. Transfer the contents of each tube to an appropriately labeled 1.5 ml microcentrifuge tube. Incubate the microcentrifuge tubes on ice for 5 min.
- Add 400 µl of MP-2 to each microcentrifuge tube. Mix the contents of each tube by gentle inversion. The mixture in the tube should turn translucent due to lysis of the bacteria. Place the tubes on ice for 5 min.
- Add 300 µl of MP-3 to each tube, and mix contents of tubes by gentle inversion. A white precipitate should appear in each tube. This precipitate contains genomic DNA and various proteins.
- Place tubes on ice for 7 min. Centrifuge the tubes at 13,000 rpm for 25 min.
- Use a pipettor to carefully transfer the supernatant from each tube (ca. 0.85 ml) to a clean microcentrifuge tube. Label tubes containing supernatants appropriately. Discard the tubes containing the pellets.
- To precipitate plasmid (BAC) DNA, add 600 µl of ice-cold isopropanol to each tube, and gently mix the contents by inversion. Place the tubes at –80ºC for 20 min.
- Spin tubes in a microcentrifuge at 13,000 rpm for 30 min. Carefully pour off the supernatants and add 1 ml of ice cold 70% ethanol to each tube. Centrifuge at 13,000 x g for 10 min.
- Gently remove and discard the supernatant from each tube. In the bottom or on the side of each tube, a small translucent pellet should be seen. This is BAC DNA. Place the microcentrifuge tubes upside down on a Kimwipe so that residual liquid can drain from the tubes. Allow the pellets to dry for 1-2 hours in a laminar-flow hood.
- Add 35 µl of sterile 1X TE to each microcentrifuge tube. Gently tap tubes to mix contents. Make sure that the pellet in each tube is covered in buffer. Place the tubes at 4ºC overnight.
- Remove tubes from the refrigerator. Gently agitate each miniprep tube by tapping on it. If pellets have not dissolved, agitate the tubes using a vortex and allow tubes to sit at room temperature for a few additional hours. Once pellets have gone into solution, proceed with step 16.
- Place 6 µl of each miniprep solution into its own 0.65 ml microcentrifuge tube. Label each of the 36 tubes with the designation of its corresponding test plate (I, II, III, or IV). The tubes will henceforth be called "NotI reaction tubes".
- Prepare a NotI digestion cocktail by placing 58.3 µl of 10X NotI buffer, 3.9 µl of 100X BSA, 268.3 µl of MBG water, and 19.4 µl of NotI in a 1.5 ml microcentrifuge tube. Mix the contents of the tube using a vortex.
- Add 9 µl of cocktail to each NotI reaction tube. Mix the contents of each reaction tube by tapping.
Note 6.2: Each reaction tube will contain 6 µl of miniprep solution, 1.5 µl 10X NotI buffer, 0.1 µl of 100X BSA, 6.9 µl of MBG water, and 0.5 µl of NotI.
- Place the reaction tubes in a microcentrifuge and spin at 10,000 rpm for 30 sec. Transfer the reaction tubes to a microcentrifuge rack. Wrap the rack in aluminum foil and place it at 37ºC for seven hours.
Note 6.3: Reaction tubes can be incubated at 37°C overnight (ca. 12-16 hrs) if necessary.
- Place 2.5 g of agarose in a 1.0 L flask. Add 0.5X TBE to a final volume of 250 ml. Place the flask on a scale, and tare the scale so the reading is 0.00 g. Remove the flask but do not reset the scale. Heat the mixture in a microwave until all of the agarose has gone into solution (this will require some boiling). Place the flask on the scale. Add MBG water until the scale once again reads 0.00 g. Cover the flask with aluminum foil, and place it in a 45°C water bath.
- Set up the large CHEF gel casting stand (see FIGURE 6.1). Place the BioRad 45-tooth comb into the comb notches nearest the end of the casting stand. Add agarose to the casting tray until it is near overflowing. Place the remaining melted agarose in a 50 ml polypropylene centrifuge tube. Place the tube in a 65°C water bath for later use. Let the gel solidify at room temperature for at least one hour.
- Carefully remove the comb from the gel.
- Take either the PFGE Lambda Ladder or one of the PFGE Midrange Ladders out of the freezer and remove the red cap from the tip of the syringe. Apply slight pressure to the plunger so that a 1.5 mm long region of agarose-embedded ladder is extruded from the syringe tip. Use a coverglass to slice this agarose from the tip of the syringe. Insert the round agarose slice into the third well of the CHEF gel (FIGURE 6.2). Place a second slice of the ladder in well 42 of the gel.
- Remove the tube containing the extra agarose from the 65°C water bath. Allow the agarose to cool to about 50°C. Carefully add melted agarose to the wells containing molecular weight ladders and plug pieces (i.e., seal the wells). Allow the melted agarose to solidify
Note 6.4: We commonly cut agarose-embedded DNA with a scalpel or razor blade. However, some prefer a coverglass arguing that nucleases may be activated by metal ions from the scalpel/razor blade.
- A BioRad CHEF Gel Apparatus is shown in FIGURE 6.3. Place 2.5 L of 0.5X TBE buffer into the electrophoresis chamber. Turn the power and the pump toggle switches on the control/power unit to the "ON" position. Turn on the cooling module, and set the temperature on the cooling module to 14°C. Allow the buffer to cool to this temperature.
- Remove the gel from the casting stand but leave it on the underlying casting plate. Wipe any agarose off the bottom of the casting plate.
- Place the plate with the gel on it in the BioRad CHEF apparatus (FIGURE 6.3). The wells should be closest to the back of the apparatus (farthest from the positive electrodes). The gel should be completely submerged in buffer.
- Unwrap the foil from the microcentrifuge rack containing the NotI digests. Add 2.0 µl of blue juice to each tube. Centrifuge the tubes at 10,000 rpm for 30 sec.
- Place the gel in the electrophoresis chamber of the BioRad CHEF apparatus.
- Using a pipettor, transfer the contents of the nine NotI reaction tubes labeled with the Roman numeral I to lanes 5-13, respectively. BE SURE TO CHANGE PIPET TIPS BETWEEN TRANSFER OF EACH SAMPLE.
- Load lanes 14-22 with the II reactions, lanes 23-31 with the III reactions, and lanes 32-40 with the IV reactions.
- Run the gel using the following parameters: volts/cm = 6.0, included angle = 120º, run time = 16 hours, initial switch time = 5 sec, final switch time = 15 sec, ramping = linear.
- Remove the gel (and underlying plate) from the CHEF apparatus. Gently remove the gel from the base plate, and place the gel in a photographic developing tray. Add enough distilled water that the gel can move freely. Add 2 drops of ethidium bromide (10 mg/ml) and place the tray on a shaker table. Set the speed of the shaker table so that the gel is not damaged, and no spilling of ethidium bromide solution occurs. Allow the gel to stain for 45 min. Carefully pour off the ethidium bromide solution (into an appropriate hazardous waste container) and add distilled water to the tray. Place the tray back on the shaker table and allow the gel to "destain" for 45 min. Place the gel on a UV light box and either photograph or digitally capture an image of the gel. A diagram illustrating the appearance of a successful transformation/miniprep/NotI digest is shown in FIGURE 6.4. Actual photographs of NotI digests of BACs can be seen in FIGURE 14.1.
INTERPRETING THE RESULTS
There are several things to consider when evaluating the results of a BAC NotI digest:
- Is ethidium bromide staining visible in the sample lanes? If so, it is likely that the minipreps were successful. If not, the miniprep DNA was most likely lost during a decanting step. The appearance of one or two empty lanes in a gel is fairly common. However, if most of the lanes are empty, it is likely that the isolation of BAC DNA needs to be performed with more care.
- Are distinct bands visible in the sample lanes? If no bands are seen, the digestion has failed (possibly due to an inactive NotI enzyme).
- The pBeloBAC11 vector has a length of 7.4 kb. If the digestion was successful, the vector should be visible as a band at 7.4 kb. The vector band should be visible in most (if not all) sample lanes.
- Do the sample lanes contain at least one band in addition to the vector band? Each lane that contains only a vector band represents a false positive. Lanes that contain a vector band and an additional band(s) are "true positives". If more than 10% of the lanes contain false positives, adjustments in the ligation and/or transformation steps are warranted.
Return to CONTENTS
Go on to CHAPTER 7